Analyses of gene activity may yield clues to roots of autism
Author Archives: Arking Lab
ASF Blog: Using the human brain to understand “omics” in autism (May 2016)
Pattern of chemical tags on DNA altered in autism brains (May 2016)
Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism (Dec 2014)
 Abstract
Abstract
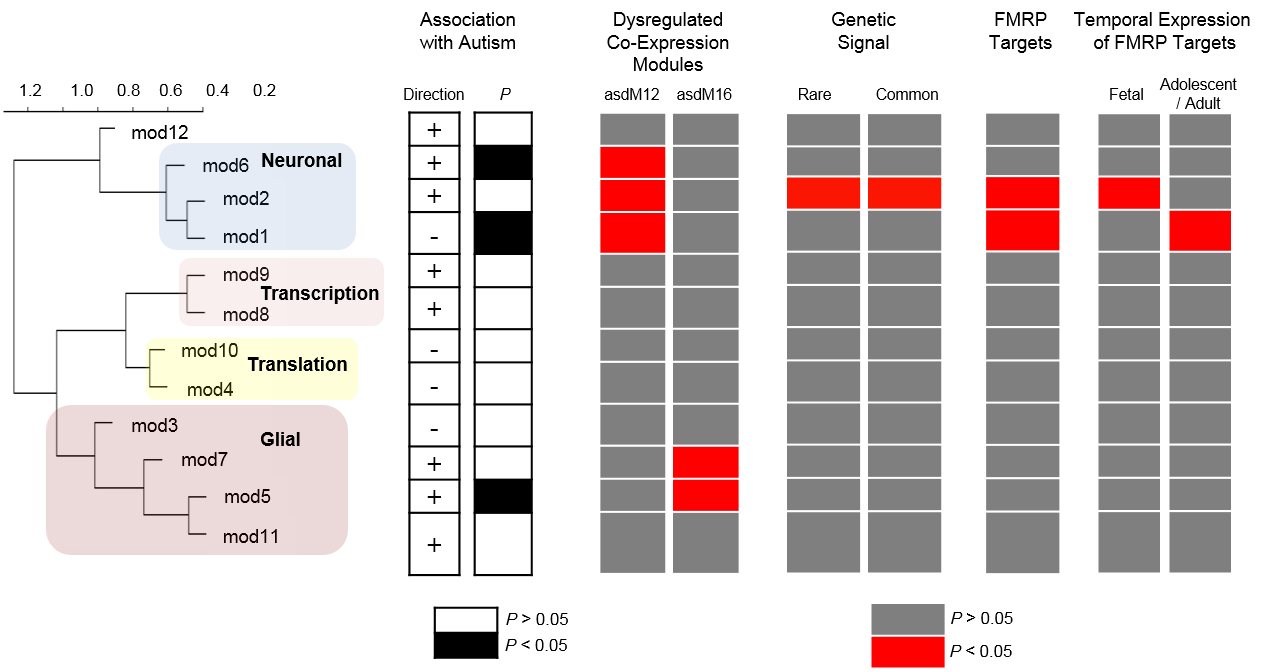
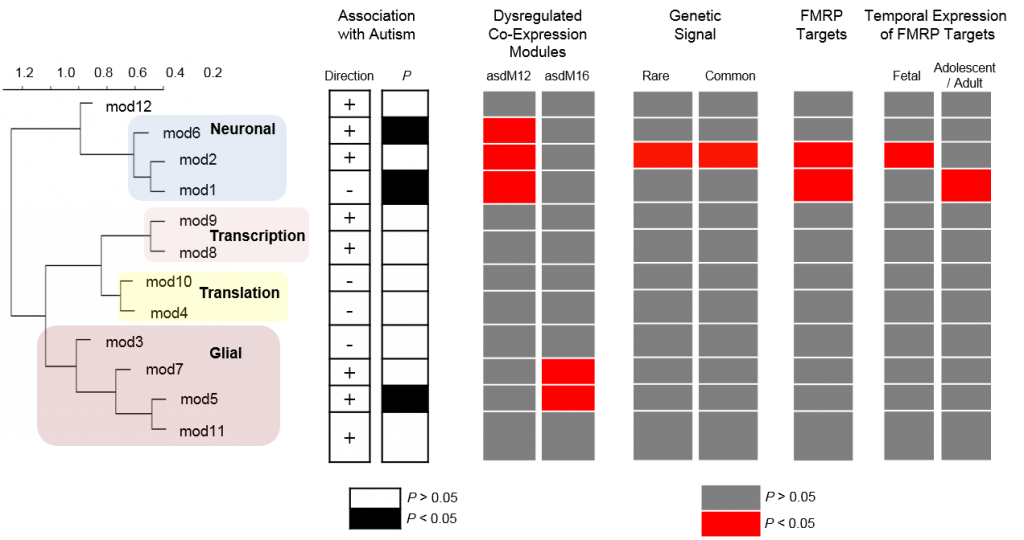
Recent studies of genomic variation associated with autism have suggested the existence of extreme heterogeneity. Large-scale transcriptomics should complement these results to identify core molecular pathways underlying autism. Here we report results from a large-scale RNA sequencing effort, utilizing region-matched autism and control brains to identify neuronal and microglial genes robustly dysregulated in autism cortical brain. Remarkably, we note that a gene expression module corresponding to M2-activation states in microglia is negatively correlated with a differentially expressed neuronal module, implicating dysregulated microglial responses in concert with altered neuronal activity-dependent genes in autism brains. These observations provide pathways and candidate genes that highlight the interplay between innate immunity and neuronal activity in the aetiology of autism.
Conclusion
We provide transcriptomic evidence for type I interferon and M2-activation state abnormalities in autism that may lead to a variety of pathologic and phenotypic consequences. We further note that there is a strong negative correlation between two differentially co-expressed modules, mod5 (activated M2-state microglia genes) and mod1 (synaptic transmission genes; r=−0.92). Recently, microglia have been identified as cells capable of restoring neural function in the ASD-model MECP2 knockout mice37. We observe, for the first time, that M2-activation state microglia genes, in particular, are altered in autism, potentially driven by type I interferon responses. This process may drive changes in neural progenitor cell proliferation and connectivity with resultant altered activity-dependent neural expression profiles in post-natal development38, 39. The linkage of this pathway to autism may lead to more accurate and predictive models of idiopathic disease that might contribute to the identification of effective therapeutic approaches.
Association of mitochondrial DNA levels with frailty and all-cause mortality (Dec 2014)
 Abstract
Abstract
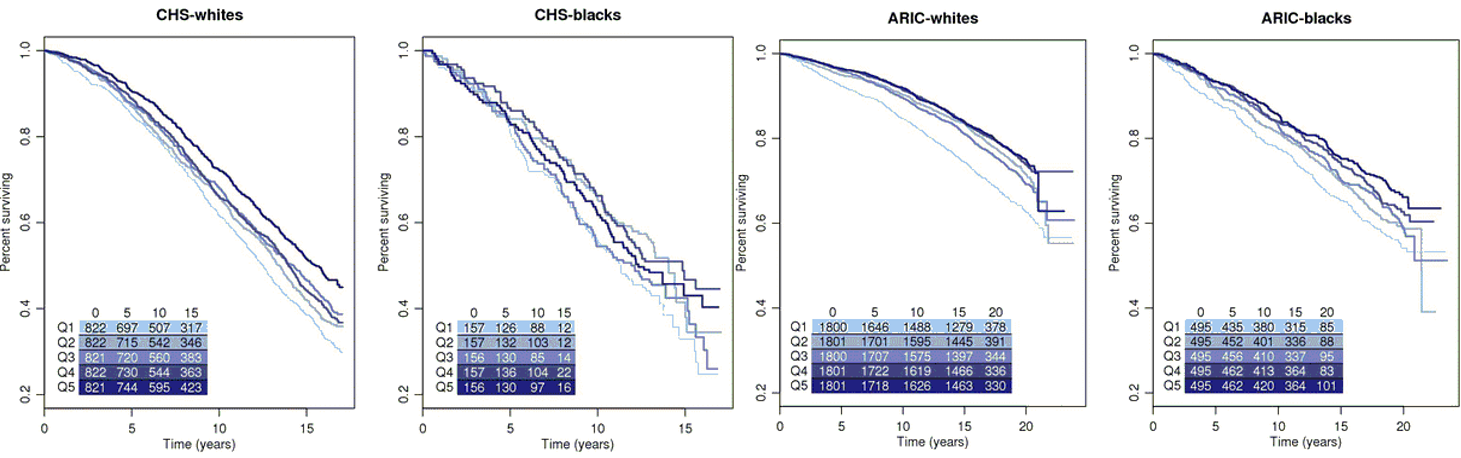
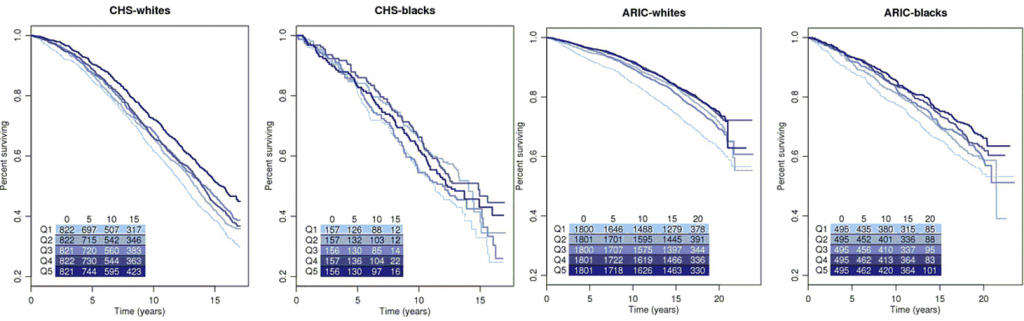
Mitochondrial function is altered with age and variants in mitochondrial DNA (mtDNA) modulate risk for several age-related disease states. However, the association of mtDNA copy number, a readily available marker which reflects mitochondrial depletion, energy reserves, and oxidative stress, on aging and mortality in the general population has not been addressed. To assess the association between mtDNA copy number and two primary outcomes—prevalent frailty and all-cause mortality—we utilize data from participants who were from two multicenter, multiethnic, community-based, prospective studies—the Cardiovascular Health Study (CHS) (1989–2006) and the Atherosclerosis Risk in Communities (ARIC) study (1987–2013). A total of 4892 participants (43.3 % men) from CHS and 11,509 participants (44.9 % men) from ARIC self-identifying as white or black were included in the analysis. mtDNA copy number, the trait of interest, was measured using a qPCR-based method in CHS and an array-based method in ARIC from DNA isolated from whole blood in participants from both cohorts. In race-stratified meta-analyses, we observe a significant inverse association of mtDNA copy number with age and higher mtDNA copy number in women relative to men. Lower mtDNA copy number was also significantly associated with prevalent frailty in white participants from CHS (OR 0.91, 95 % CI 0.85–0.97). Additionally, mtDNA copy number was a strong independent predictor of all-cause mortality in an age- and sex-adjusted, race-stratified analysis of 16,401 participants from both cohorts with a pooled hazard ratio of 1.47 (95 % CI 1.33–1.62) for the lowest quintile of mtDNA copy number relative to the highest quintile.
Key messages
- mtDNA copy number is associated with age and sex
- Lower mtDNA copy number is also associated with prevalent frailty
- mtDNA copy number is a significant predictor of all-cause mortality in a multiethnic population
RNA-Seq optimization with eQTL gold standards (Dec 2013)
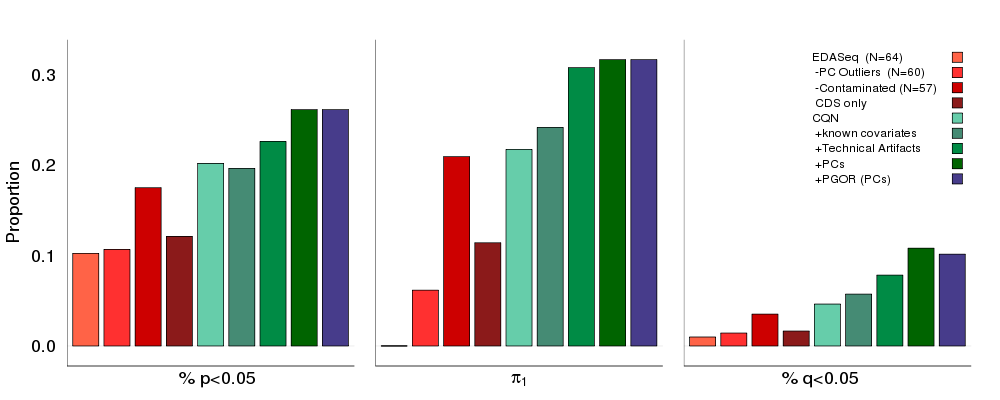 Background
Background
RNA-Sequencing (RNA-Seq) experiments have been optimized for library preparation, mapping, and gene expression estimation. These methods, however, have revealed weaknesses in the next stages of analysis of differential expression, with results sensitive to systematic sample stratification or, in more extreme cases, to outliers. Further, a method to assess normalization and adjustment measures imposed on the data is lacking.
Results
To address these issues, we utilize previously published eQTLs as a novel gold standard at the center of a framework that integrates DNA genotypes and RNA-Seq data to optimize analysis and aid in the understanding of genetic variation and gene expression. After detecting sample contamination and sequencing outliers in RNA-Seq data, a set of previously published brain eQTLs was used to determine if sample outlier removal was appropriate. Improved replication of known eQTLs supported removal of these samples in downstream analyses. eQTL replication was further employed to assess normalization methods, covariate inclusion, and gene annotation. This method was validated in an independent RNA-Seq blood data set from the GTEx project and a tissue-appropriate set of eQTLs. eQTL replication in both data sets highlights the necessity of accounting for unknown covariates in RNA-Seq data analysis.
Conclusion
As each RNA-Seq experiment is unique with its own experiment-specific limitations, we offer an easily-implementable method that uses the replication of known eQTLs to guide each step in one’s data analysis pipeline. In the two data sets presented herein, we highlight not only the necessity of careful outlier detection but also the need to account for unknown covariates in RNA-Seq experiments.